Machine learning interatomic potential
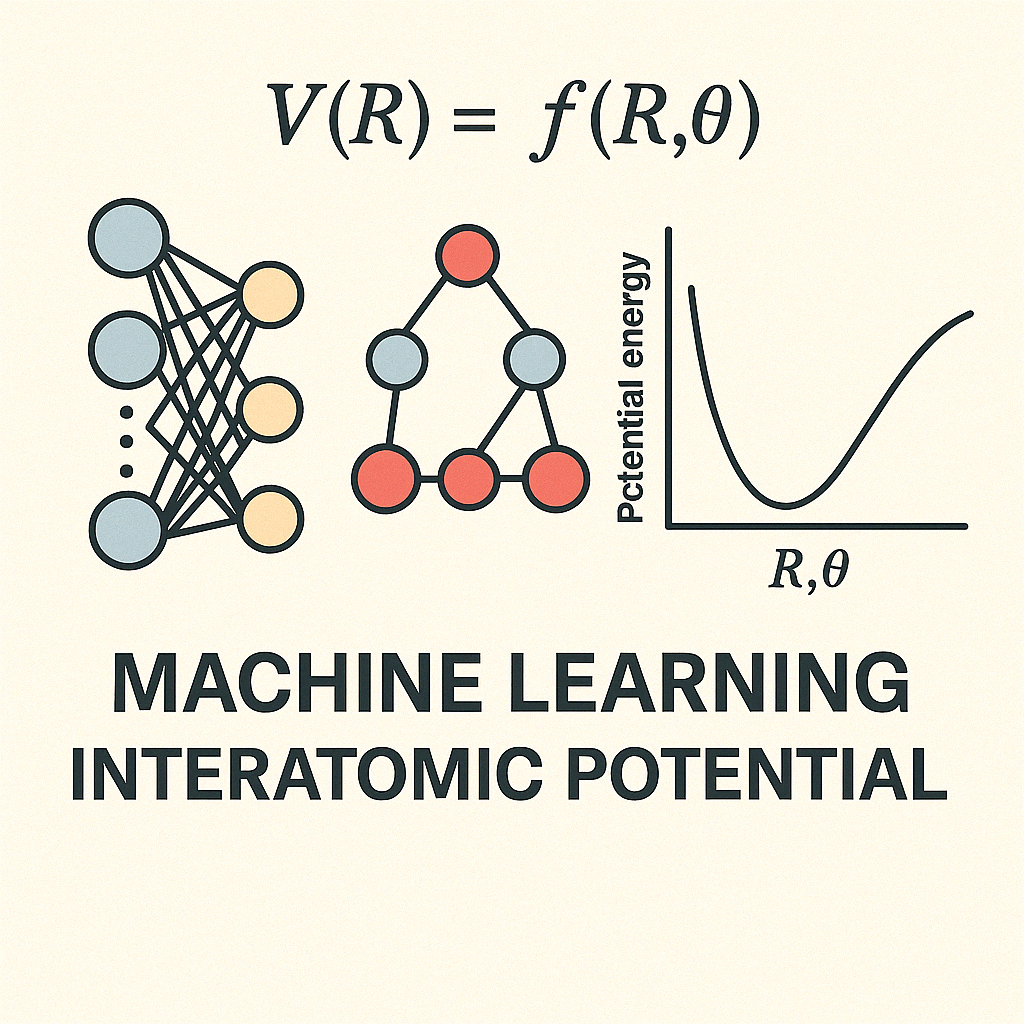
Machine Learning Interatomic Potentials for Materials Science
Understanding and predicting the behavior of materials at the atomic level is essential for designing novel materials with targeted properties. Traditional atomistic simulation methods—such as those based on quantum mechanics (e.g., density functional theory)—offer high accuracy but are computationally expensive and thus limited in scale and time. On the other hand, classical empirical potentials are computationally efficient but often lack the precision required to capture complex atomic interactions. To bridge this gap, our research focuses on the development and application of machine learning interatomic potentials (MLIPs), which aim to combine the accuracy of quantum mechanical methods with the efficiency of classical potentials.
MLIPs use data-driven models trained on high-fidelity quantum mechanical datasets to learn the underlying physics of atomic interactions. By leveraging advanced machine learning algorithms—including neural networks, Gaussian processes, and graph-based architectures—these potentials can accurately capture a wide range of interatomic forces and energies across diverse materials systems. Our group is actively developing robust, transferable MLIP frameworks tailored for various materials, including metals, oxides, and hybrid systems, with applications spanning energy storage, catalysis, and electronic materials.
Beyond development, we also focus on integrating MLIPs into large-scale atomistic simulations to uncover new insights into the dynamic, thermodynamic, and mechanical behavior of complex materials. Through this effort, we aim to enable accelerated materials discovery and design by drastically reducing the computational cost while maintaining high accuracy. Our work contributes to advancing computational materials science into the era of data-driven modeling, and aligns with the broader vision of autonomous materials research.
-
Machine Learning-Enabled Exploration of the Electrochemical Stability of Real-Scale Metallic Nanoparticles
Nature Communications 14:3004 (2023)
Machine Learning Filters Out Efficient Electrocatalysts in the Massive Ternary Alloy Space for Fuel Cells
Applied Catalysis B: Environmental 339, 123128 (2023)